
Source: C2S-Scale – Scaling LLMs for Next‑Generation Single‑Cell Analysis (Preprint, Oct 10, 2025)
Read Official Blog: https://blog.google/technology/ai/google-gemma-ai-cancer-therapy-discovery/
Listen in many languages:
Executive summary
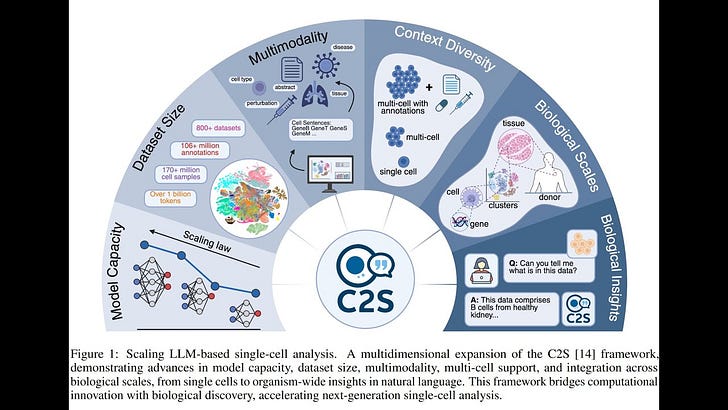
- What it is: C2S-Scale is a family of Large Language Models (LLMs) trained to analyze single-cell RNA-seq by converting gene-expression profiles into textual “cell sentences” and jointly learning from >50 million cells plus biological text and metadata.
- Why it matters: It unifies transcriptomic and textual data at scale, enabling predictive, generative, and natural-language interpretation tasks in a single model, including multi-cell spatial reasoning and virtual perturbation screens.
- Headline finding: Scaling to 27B parameters and aligning with reinforcement learning produces consistent gains across prediction, generation, and reasoning, and directly enabled a dual-context virtual screen that predicted—and wet-lab validated—silmitasertib as an interferon‑conditional amplifier of antigen presentation.
- Availability: Code (github.com/vandijklab/cell2sentence) and model weights (Hugging Face). Preprint, not peer reviewed.
- See it in action at Hugging Face Spaces - : https://huggingface.co/spaces/napoles3d/demo_C2S_Scale
Main themes
- LLMs for biology via data engineering, not bespoke architectures: transform scRNA-seq to “cell sentences” so standard LLMs can ingest transcriptomes alongside text.
- Scaling laws apply: performance steadily improves from 410M to 27B parameters across diverse single-cell tasks.
- Multimodality and multi-cell context: integrates gene expression, metadata, literature text; supports multi-cell prompts for spatial inference and dataset-level interpretation.
- Alignment for biology: reinforcement learning (GRPO) with biology-aware rewards improves question answering and perturbation prediction.
- From analysis to discovery: virtual screening with context-specific criteria leading to testable, validated hypotheses.
How it works (method in brief)
- Cell2Sentence (C2S): For each cell, rank-order genes by expression and emit the top K as a text sequence of gene names (“cell sentence”). The rank-to-expression mapping is reversible with low information loss (R2 ~0.85 in examples).
- Pretraining corpus: >50M human/mouse cells from CELLxGENE and HCA, metadata, associated papers; natural language plus transcriptomic “tokens” exceed 1B tokens.
- Models and training: Decoder-only Transformer LLMs (Gemma 2/Pythia backbones), 410M–27B params. Pretrained with next-token prediction across multi-task prompts; fine-tuned with SFT and GRPO. Parameter-efficient LoRA supported.
- New metric: scFID (single-cell Fréchet Inception Distance), an adaptation of FID using single-cell foundation model embeddings to assess fidelity of generated cells at the distribution level.
Key findings and capabilities
- Broad task coverage in one model:
- Prediction: cell type, tissue, dataset interpretation, question answering, spatial neighborhood inference, single-cell vs bulk matching.
- Generation: conditional/unconditional single-cell and multi-cell generation; perturbation response prediction (drugs, cytokines, genetic knockouts).
- Natural-language interpretation: cluster captioning and dataset abstracts; generalizes to unseen studies.
- Scaling improves performance:
- Larger models consistently outperform smaller ones across cell type annotation, dataset interpretation, and conditional generation; gains hold under full and parameter-efficient fine-tuning; more data also improves results.
- Natural-language interpretation across scales:
- Cells: immune dataset cell type classification in natural language reached 95.43% accuracy (vs scGPT 93.1%, Geneformer 94.0%).
- Clusters: outperforms Llama, Gemini, GPT‑4o, and other biomedical LLMs on cluster captioning by BioBERTScore.
- Datasets: produces high-quality abstracts for held-out and out-of-distribution datasets; best BERTScore among evaluated models (Llama, Meditron, BioMistral, Gemini, GPT‑4o).
- Spatial reasoning from multi-cell context:
- Predicts spatial neighborhood membership better than scGPT and GPT‑4o.
- Incorporating gene interaction knowledge (CellPhoneDB, BioGRID) as natural-language prompts further improves spatial prediction; combining both sources yields the largest gain.
- Question answering with RL:
- scQA dataset (~2k Q/A pairs) built from manuscripts and sampled scRNA-seq data.
- C2S-Scale SFT surpasses state-of-the-art LLMs; GRPO with BioBERTScore reward further improves answer quality.
- Perturbation response prediction:
- Dong et al. cytokines: best performance vs scGen, CellOT, scGPT on unseen combinations; captures nonlinear synergies; GRPO reduces scFID on IFN genes by 16% and improves MMD/Wasserstein scores.
- L1000 compounds: GRPO improves apoptosis-program accuracy (Kendall’s tau +9.2% for 410M, +4.9% for 1B; Pearson’s r +6.6% and +3.6% respectively).
- scFID provides stable, structure-aware distributional evaluation; complements Kendall’s tau/Pearson correlation on genes.
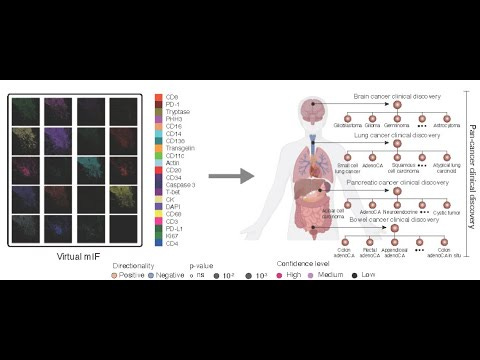
- Virtual screen and wet-lab validation:
- Dual-context strategy (immune-context-positive vs -neutral) for MHC‑I program upregulation.
- Model predicted a strong context split for silmitasertib (CX‑4945, CK2 inhibitor): robust effect under low IFN, negligible effect without IFN.
- Validation in human neuroendocrine models unseen in training:
- Merkel cell origin: no effect alone; with low-dose IFN‑β, MHC‑I MFI increased 13.6% (10 nM) and 34.9% (1000 nM).
- IFN‑γ also amplified: +24.9% (10 nM), +37.3% (300 nM).
- Pulmonary model: +17.1% (10 nM), +28.1% (100 nM).
- Interpretation: silmitasertib behaves as an interferon-conditional amplifier, lowering the response threshold for STAT1/IRF1-driven MHC‑I upregulation.
Benchmark highlights (selected)
- Cell type annotation (immune): 95.43% vs scGPT 93.1%, Geneformer 94.0%.
- Natural-language QA: +3% BERTScore vs GPT‑4o; GRPO further improves.
- Spatial neighborhood prediction: surpasses scGPT and GPT‑4o; gene-interaction prompts (CellPhoneDB, BioGRID) add significant gains.
- Perturbation (Dong et al.): best MMD, Wasserstein, scFID across held-out single and combinatorial cytokine conditions; GRPO yields additional gains.
- L1000 apoptosis: RL improves rank- and value-based metrics as noted above.
Applications in business
- Pharma and biotech R&D
- Virtual perturbation screening at scale across cell types and contexts; prioritize compounds with context-conditional effects.
- Target/pathway discovery from natural-language interpretation of scRNA-seq and literature.
- Mechanism-of-action hypotheses via multi-cell, spatial, and receptor–ligand reasoning.
- Biomarker discovery and patient stratification by tissue, cell state, or immune context.
- CROs and translational research
- Augment experimental design with in-silico previews; focus wet-lab spend on high-yield conditions.
- Automated reporting: cluster/dataset captioning and study summaries for client deliverables.
- Diagnostics and precision medicine
- Interpretable single-cell signatures; context-aware risk scoring and IFN/MHC‑I pathway status for immunotherapy readiness.
- Life-science software and data platforms
- LLM-native single-cell analytics: embedding, annotation, spatial inference, and dataset summarization exposed via APIs.
- Knowledge integration: unify omics, metadata, and literature into searchable, explainable outputs.
- Cloud/AI vendors
- Domain-specialized LLM service for single-cell analysis with parameter-efficient fine-tuning for customer data.
- Investors and strategy
- Signals a path from single-cell modeling to actionable, validated hypotheses; potential moat via multimodal corpora, RL alignment, and domain evaluation (scFID, scQA).
Deployment and integration
- Inputs: scRNA-seq (single-cell, multi-cell contexts), bulk RNA-seq (generalizes), metadata, abstracts/papers, gene interaction text.
- Outputs: labels (cell type/tissue), cell sentences (generated cells), embeddings, captions/abstracts, Q/A, perturbation predictions.
- Footprint: 410M–27B params; LoRA for efficient adaptation; trained on GPUs/TPUs; open weights on HF; code on GitHub.
- Data sources leveraged: CELLxGENE, Human Cell Atlas, CellPhoneDB, BioGRID, L1000, CosMx liver, pan-cancer bulk RNA-seq, selected GEO datasets.
Limitations and open questions
- Preprint status: not peer reviewed; some benchmarks use model-generated captions/abstract variants; scQA constructed with GPT-4.5 assistance.
- Representation limits: rank-based “cell sentence” compresses expression; authors report low information loss with dataset-calibrated reverse mapping.
- Generalization: strongest validation shown for immune/IFN contexts and selected cell types; broader tissue and clinical generalization remains to be established.
- Reward design: RL gains depend on chosen gene programs (e.g., IFN/apoptosis); transferability to other pathways requires careful reward curation.
- Safety and ethics: model outputs should not be used for clinical decision-making without validation; avoid over-reliance on literature priors embedded in LLMs.
Key quotes from the text
- “C2S-Scale unifies transcriptomic and textual data at unprecedented scales, surpassing both specialized single-cell models and general-purpose LLMs to provide a platform for next-generation single-cell analysis and the development of ‘virtual cells.’”
- “Scaling the model to 27 billion parameters yields consistent improvements in predictive and generative capabilities and supports advanced downstream tasks that require synthesis of information across multi-cellular contexts.”
- “We also introduce a novel metric, single-cell Fréchet Inception Distance (scFID), for assessing generative performance.”
- “C2S-Scale outperforms both transcriptomic and natural language foundation models across diverse predictive and generative single-cell tasks.”
- “Fine-tuning with gene interaction data further improves C2S-Scale’s ability to predict spatial relationships.”
- “Following GRPO training, C2S-Scale significantly outperforms the SFT baseline on the scQA dataset.”
- “C2S-Scale outperforms existing methods on the Dong et al. dataset… capturing nonlinear synergistic effects.”
- “The screen revealed a pronounced context split for the kinase inhibitor silmitasertib… suggesting its potential as a synergistic, interferon-conditional amplifier of antigen presentation.”
- “Experimental validation in human cell models unseen during training confirmed this hypothesis.”
What’s novel vs prior art
- Uses off-the-shelf LLMs with a reversible textual transformation of scRNA-seq, avoiding custom architectures while enabling seamless integration of literature and metadata.
- Demonstrates consistent scaling laws for biological reasoning with LLMs, up to 27B parameters.
- Introduces scFID for distribution-level evaluation of generated cells.
- Shows multi-cell and spatial reasoning emerging from context prompts and enhanced by receptor–ligand knowledge in text form.
- Validates a model-derived, context-conditional drug effect experimentally in previously unseen cell models.
Suggested next steps for organizations
- Run a proof-of-concept: annotate datasets, generate cluster captions and dataset abstracts; compare to current pipelines.
- Evaluate virtual perturbation predictions in a domain of interest (e.g., cytokines, chemotherapies) and test a short list in vitro.
- Pilot multi-cell spatial reasoning on in-house spatial datasets; enrich with internal interaction knowledgebases in natural-language form.
- Design RL rewards aligned to your therapeutic area (e.g., EMT, metabolic rewiring) and fine-tune with GRPO.
- Integrate embeddings and natural-language interfaces into knowledge discovery tools; assess gains in scientist productivity.
Provenance and availability
- Institutions: Yale University, Google Research, Google DeepMind, USC, Brown University.
- Code: https://github.com/vandijklab/cell2sentence
- Weights: Hugging Face (C2S-Scale family; sizes up to 27B).
- Data used: CELLxGENE, Human Cell Atlas, CosMx liver spatial data, interaction databases (CellPhoneDB, BioGRID), L1000, Dong et al. cytokine dataset, pan-cancer bulk data, selected GEO datasets.
Contact and correspondence (from preprint)
- Corresponding authors: jeffrey.ishizuka@yale.edu, shekazizi@google.com, david.vandijk@yale.edu
One-line takeaway
By reframing single-cell expression as language, C2S-Scale turns state-of-the-art LLMs into multimodal, scalable engines for single-cell analysis, enabling accurate prediction, natural-language interpretation, spatial reasoning, and validated, context-aware therapeutic discovery.
Created with AI